Los científicos llevan mucho tiempo intrigados por cómo los glóbulos rojos en maduración logran producir toda la hemoglobina necesaria para transportar oxígeno a los tejidos, incluso después de desprenderse de las estructuras vitales para su producción. Ahora, una nueva investigación de la Facultad de Medicina de la Universidad de Maryland (UMSOM) ha encontrado una pieza clave: en las etapas finales de su maduración, los glóbulos rojos pueden importar hemo, un componente crucial de la hemoglobina que contiene hierro, de otras células. Este sorprendente hallazgo no solo refuta suposiciones arraigadas sobre el hemo y los glóbulos rojos, sino que podría conducir a nuevas terapias para trastornos sanguíneos genéticos hereditarios como la β-talasemia.
En el estudio publicado hoy en la revista Science , investigadores de la UMSOM demostraron por primera vez que, bajo ciertas condiciones, los glóbulos rojos inmaduros, conocidos como eritroblastos, pueden importar hemo de las células circundantes incluso después de desechar sus propias mitocondrias productoras de hemo. Lo hacen a través de una proteína transportadora conocida como gen 1 sensible al hemo (HRG1), que el laboratorio del Dr. Hamza descubrió años atrás en un gusano microscópico sin sangre.
“Hemos demostrado que esta proteína transportadora, HRG1, es esencial para la producción de glóbulos rojos sanos y maduros, especialmente cuando el cuerpo necesita producirlos rápidamente debido a ciertas situaciones de estrés, como la falta de oxígeno a gran altitud o durante una hemorragia”, afirmó el investigador principal del estudio, Iqbal Hamza, PhD , profesor de Pediatría de la Facultad de Medicina de la Universidad de Maryland (UMSOM) en el Centro para el Transporte de Oxígeno en la Sangre y la Hemostasia ( CBOTH ). “Descubrimos que, en ausencia de HRG1, los glóbulos rojos que se producen son subóptimos, lo que significa que cuando el sistema está bajo estrés y debe producir más glóbulos rojos de lo habitual, puede tener dificultades para hacerlo sin HRG1”.
Este descubrimiento tiene relevancia clínica en el contexto de la anemia por deficiencia de hierro, el trastorno nutricional más frecuente en el mundo. «Una característica común de la anemia son los glóbulos rojos pálidos y con deficiencia de hemoglobina», afirmó el Dr. Hamza, profesor
de Ciencias Animales y Avícolas en la Universidad de Maryland, College Park. «Al identificar estrategias para mitigar la anemia, como la regulación del suministro de hemo a través de HRG1 para mejorar la producción de hemoglobina, podríamos reducir sustancialmente la morbilidad y la mortalidad asociadas a esta afección».
El enigma de la hemoglobina
La investigación intentó dilucidar cómo los glóbulos rojos en maduración satisfacen los extraordinarios requerimientos de hemo necesarios para seguir produciendo hemoglobina en las etapas finales de su maduración tras la pérdida de sus mitocondrias. «Intentábamos identificar la vía metabólica implicada en este proceso», explicó el Dr. Hamza.
Él y sus colegas plantearon la hipótesis de que estos glóbulos rojos en desarrollo importan hemo a través de HRG1 para proporcionar un impulso final a sus niveles de hemoglobina.
Para comprobar su hipótesis, el Dr. Hamza y su equipo utilizaron primero la secuenciación de ARN de células individuales en glóbulos rojos inmaduros de ratones para confirmar que el gen HRG1 se expresa en los glóbulos rojos en desarrollo y que su expresión aumentaba a medida que las células maduraban. Posteriormente, emplearon un modelo animal para estudiar qué ocurría al inactivar el gen HRG1.
Los investigadores descubrieron que los ratones con el gen HRG1 inactivado no podían aumentar adecuadamente su producción de glóbulos rojos y desarrollaban anemia cuando se les sometía a estrés y se les obligaba a generar más glóbulos rojos. Los ratones normales, en cambio, eran capaces de reponer sus glóbulos rojos.
Posteriormente, los investigadores utilizaron citometría de flujo para demostrar que los glóbulos rojos producidos por los ratones modificados genéticamente no acumulaban suficiente hemoglobina y morían antes de alcanzar la madurez completa, lo que indica que la proteína HRG1 era esencial para que las células acumularan suficiente hemo para sobrevivir y funcionar correctamente.
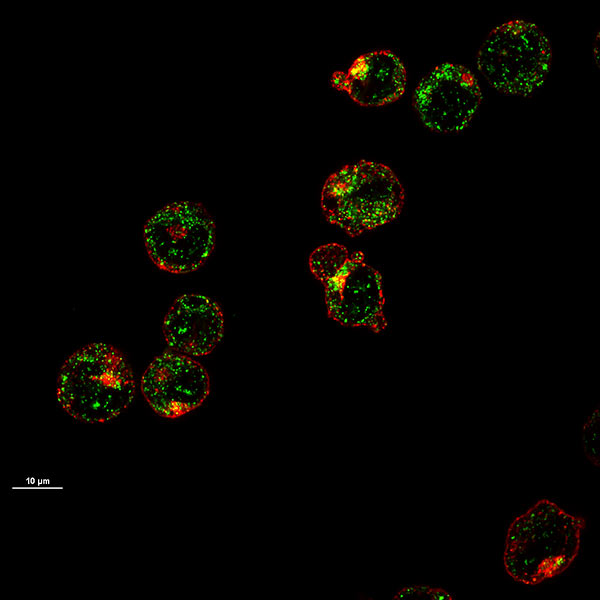
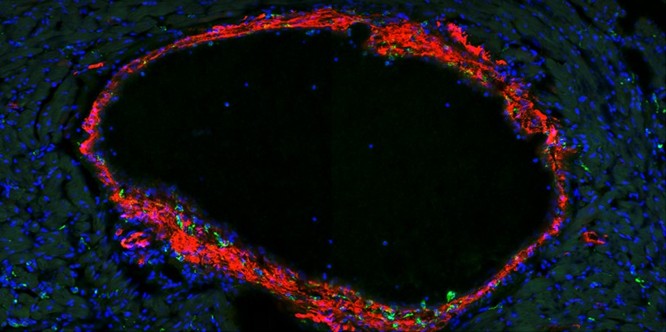
Los eritrocitos precursores en maduración muestran transportadores del gen 1 sensible al hemo (HRG1) (en verde). Estos transportadores, expresados en las membranas celulares, trasladan el hemo al interior de la célula para la síntesis de hemoglobina.
Implicaciones para el tratamiento de la β-talasemia
El Dr. Hamza y su equipo también examinaron un modelo de ratón con β-talasemia, un trastorno sanguíneo genético hereditario que causa una producción insuficiente de hemoglobina y glóbulos rojos, lo que provoca anemia grave. Dado que este trastorno sanguíneo, que se estima afecta a 10 millones de personas en todo el mundo, se caracteriza por la acumulación de hemo libre en exceso que no se integra a la hemoglobina, los investigadores plantearon la hipótesis de que reducir la actividad del gen HRG1 podría limitar la acumulación de hemo libre tóxico y mitigar los síntomas. Para comprobarlo, eliminaron una copia del gen HRG1 en el modelo animal de β-talasemia y observaron mejoras significativas en la producción de glóbulos rojos y en la anemia.
“Este trabajo revela una vía de transferencia intercelular de hemo previamente desconocida que ayuda a mantener la producción de glóbulos rojos bajo estrés”, afirmó Mark T. Gladwin, MD , Decano de la Facultad de Medicina de la Universidad de Maryland, Vicepresidente de Asuntos Médicos de la Universidad de Maryland en Baltimore y Profesor Distinguido John Z. y Akiko K. Bowers. “Las implicaciones se extienden a un amplio espectro de trastornos sanguíneos, incluyendo la anemia falciforme y la β-talasemia, donde el desequilibrio del hemo provoca inflamación, estrés oxidativo y daño orgánico. La identificación de HRG1 como regulador de la disponibilidad de hemo abre interesantes posibilidades terapéuticas para afecciones en las que el cuerpo tiene dificultades para mantener una producción saludable de glóbulos rojos”.
El laboratorio del Dr. Hamza está llevando a cabo nuevos experimentos para determinar la importancia relativa del hemo producido por las mitocondrias frente al hemo importado por HRG1 en diferentes enfermedades, incluida la anemia por deficiencia de hierro. Su laboratorio también recibió recientemente una prestigiosa subvención del Programa de Ciencias de la Frontera Humana para investigar cómo se propaga el hemo a través de las células después de ser producido en las mitocondrias.
“Esperamos dilucidar cómo se transporta, se distribuye y se inserta el hemo en las proteínas”, dijo el Dr. Hamza. “Esto requerirá desarrollar métodos para finalmente ‘ver’ el hemo dentro de las células individuales”.
Más adelante, su objetivo es buscar nuevas dianas farmacológicas que puedan regular y ajustar la cantidad de HRG1 que se expresa, lo que podría proporcionar una nueva herramienta para tratar la β-talasemia y otros trastornos sanguíneos genéticos hereditarios, así como la anemia.
El Dr. Hamza realizó el estudio junto con sus colegas de la Facultad de Medicina de la Universidad de Maryland (UMSOM) , Audrey Belot, PhD , Xiaojing Yuan, PhD , Satoru Otsuru, MD, PhD y Amaury Maros, así como con los estudiantes de posgrado Andrew Rock y Sohini Dutt, y la asistente de investigación del laboratorio de Hamza, Gia Haemmerle. David Bodine, PhD, jefe de la División de Genética y Biología Molecular y director de la Sección de Hematopoyesis del Instituto Nacional de Investigación del Genoma Humano, también fue coautor del estudio.
Fuente: Jon Kelvey/ University of Maryland/ medschool.umaryland.edu